







B. REPLACER LE PATIENT AU CoeUR DE L'INNOVATION
1. Lever les freins dans l'accès précoce aux innovations
a) Des délais d'accès encore trop longs
• Une préoccupation forte des acteurs du secteur est de permettre un accès rapide des patients à l'innovation . En matière pharmaceutique, l'Union européenne 50 ( * ) a fixé à 180 jours le délai de référence pour l'accès au marché . Ce délai correspond à la durée considérée comme le maximum pouvant séparer l'autorisation de mise sur le marché et la prise en charge du médicament.
La loi reprend ce délai d'accès, fixé à 180 jours pour la ville comme pour la liste en sus, et à 75 jours dans le cas d'une rétrocession. Le respect de ces délais était l'un des engagements du Gouvernement à l'issue du 8 e CSIS.
Le ministère de la recherche a souligné sur ce sujet ce qu'il considère comme « un problème structurel majeur » : les autorisations de mise sur le marché (AMM) des médicaments sont attribuées par l'Agence européenne du médicament (« European Medicines Agency » - EMA), permettant la commercialisation sur tout le territoire européen sans frontières internes, dans le cadre du marché commun, alors que la fixation des prix des médicaments est une compétence étatique , amenant à des disparités intra-européennes en termes de prix et de réglementations. Dans les faits, comme le précise le ministère, il peut exister en France un délai de plusieurs années entre l'autorisation de mise sur le marché obtenu à l'EMA et l'autorisation de vente du produit qui dépend des négociations au CEPS.
Les acteurs du secteur pharmaceutique ont tous regretté des délais encore très longs d'accès au médicament, même si la situation tend à s'améliorer sur les dernières années.
• Selon le CEPS 51 ( * ) , en moyenne, le délai de 180 jours a été respecté en 2019 pour les inscriptions en ville et sur la liste en sus, avec respectivement 144 et 147 jours. Cependant, ces délais moyens sont plus faibles pour les génériques et biosimilaires et plus longs pour les produits issus des ATU et post-ATU.
Derrière ces délais moyens, de grandes disparités apparaissent, avec des délais beaucoup plus longs en ce qui concerne les médicaments innovants.
Ainsi, selon les études réalisées par le Leem 52 ( * ) :
- le délai moyen d'évaluation par la HAS était de 168 jours en moyenne en 2020 selon la base de données Prioritis ;
- le délai moyen de négociation de prix et de publication au Journal officiel était de 229 jours pour les nouvelles spécialités (hors génériques et biosimilaires) en 2019 (dont 62 jours de publication au Journal officiel ).
Concernant les nouveaux médicaments ayant reçu une première AMM, l'étude européenne réalisée par les industries pharmaceutiques estime à 527 jours en 2020 le délai moyen d'accès en France 53 ( * ) . Il est très difficile d'estimer finement le délai d'accès selon l'amélioration du service médical rendu (ASMR) et le délai réel d'accès pour les médicaments innovants en primo-inscription. Le Leem propose une meilleure identification, dans ces délais moyens, des médicaments innovants, notamment ceux bénéficiant d'une présomption d'innovation reconnue par l'Agence européenne des médicaments, mais aussi ceux bénéficiant d'une autorisation temporaire d'utilisation (ATU) : considérer l'ATU comme un délai nul ferait chuter le délai moyen à 257 jours .
Selon les industriels, la France obtient la 21 e place sur 28 pays en termes de rapidité d'accès aux molécules avec un délai moyen de disponibilité de la molécule pour le patient français de 15 à 18 mois.
• La HAS a présenté en janvier 2020 un plan d'action pour les médicaments innovants, autour de trois axes :
- rendre des avis conditionnels, le temps de lever les incertitudes ;
- suivre les médicaments en vie réelle pour vérifier les promesses initiales ;
- renforcer l'agilité de la HAS pour mieux accompagner l'innovation. Sur ce dernier point, la HAS entend principalement se concentrer sur des évaluations à forte valeur ajoutée mais aussi promouvoir les procédures d'évaluation accélérées (dites de « fast-track »).
Le bilan intermédiaire de la HAS est jugé positif avec une montée en charge du dispositif d'évaluation anticipée qui a montré des résultats sur les délais avec un délai moyen de traitement passé de 126 à 105 jours entre 2019 et 2020 pour une proportion de dossiers ayant une évaluation complète qui a augmenté pour atteindre 68 % en 2020. Concernant la délivrance d'évaluations conditionnelles pour certains médicaments prometteurs dans des maladies graves en situation de besoin médical non couvert avec réévaluation, cette possibilité a été mise en oeuvre pour trois spécialités dont l'une a vu son ASMR réévaluée de IV à III au vu du gain démontré pour la survie des patients.
Des marges de progression sont en outre identifiées par la HAS qui déplore une sous-mobilisation de deux dispositifs :
- le pré-dépôt , qui n'a concerné en 2020 que 14 % des dossiers de primo-inscription et d'extension d'indication en procédure d'instruction complète, alors que ce processus a montré une réduction du délai à une moyenne de 78 jours ;
- la présomption d'innovation . Ce processus se distingue d'une évaluation, il s'agit d'une reconnaissance permettant d'obtenir une instruction anticipée dès la demande d'AMM. En 2020, seulement sept industriels l'ont sollicité pour dix médicaments.
Proposition n° 20 : améliorer le traitement des dossiers d'évaluation en vue de la prise en charge afin de garantir un accès rapide aux innovations.
b) Un cadre de l'accès précoce rénové
À la suite du CSIS de 2018, différentes adaptations réglementaires ont été apportées aux dispositifs français d'accès précoce, particulièrement lors de la LFSS pour 2019 avec la possibilité de délivrer des autorisations temporaires d'utilisation aux extensions d'indication et lors de la loi de financement de la sécurité sociale (LFSS) pour 2021, avec deux dispositifs : l'accès précoce et l'accès compassionnel . Cette refonte vise à clarifier les circuits d'accès dérogatoires en les réorganisant non plus d'après leur seule dénomination, mais d'après leur finalité. Il réunit l'ATU et la recommandation temporaire d'utilisation (RTU) sous trois régimes dérogatoires : accès précoce, accès compassionnel et prescription « hors AMM ».
L'ANSM considère que le dispositif adopté fin 2018 a permis un accès précoce dans des situations d'absence d'alternative satisfaisante en oncologie. Ainsi, dix-sept ATU de cohorte (ATUc) d'extension ont été autorisées en 2020, en plus des 20 ATUc octroyées avant l'AMM.
Une réforme de l'accès précoce en cours de mise en oeuvre
La loi de financement pour 2021 54 ( * ) a redéfini les dispositifs d'accès précoce, supprimant les ATU et RTU au profit de deux dispositifs nouveaux : l'accès précoce pour les médicaments innovants en développement et l'accès compassionnel quand le développement n'est pas envisagé.
Ces modifications poursuivent trois objectifs :
- répondre aux besoins d'accès aux médicaments couverts par les dispositifs actuels ;
- être plus homogène, simple et lisible pour les acteurs ;
- offrir des garanties par rapport à la soutenabilité de notre système de santé.
Cette réforme doit entrer en vigueur au 1 er juillet 2021. Elle modifie profondément la répartition des rôles entre autorités sanitaires en donnant à la HAS un pouvoir décisionnel dont elle était jusqu'ici privée. La HAS est, avec la réforme, chargée de l'autorisation des accès précoces, sur avis conforme de l'ANSM.
L'accès précoce issu de la réforme prévoit également un suivi en vie réelle avec un recueil prévu de données sur l'efficacité, les effets indésirables et les conditions réelles d'utilisation.
• L'ANSM estime que cette réforme va permettre de fluidifier les processus d'évaluation menée dans les différentes institutions (ANSM, HAS, direction de la sécurité sociale) en les rassemblant dans la même unité de temps en vue d'atteindre un accès plus rapide aux traitements et de simplifier les démarches des laboratoires, via notamment le dossier commun ANSM/HAS, sur un portail commun . Cependant, elle admet que l'efficacité du nouveau dispositif dépendra de la compréhension et de la lisibilité qu'en auront les laboratoires .
• Ce dispositif nouveau est également salué par les entreprises du médicament, qui estiment le nouveau système de financement et de prise en charge comme un modèle correspondant à leurs attentes et porteur d'une plus grande lisibilité pour les acteurs.
L'ANSM a veillé au cours des dernières années à rendre plus lisible et accessible l'accès aux produits innovants. Elle a notamment mis en place en 2019 un référentiel des spécialités disponibles en ATU nominatives (ATUn) afin d'améliorer la connaissance des prescripteurs sur la délivrance possible des molécules. Une hausse sensible du nombre de nouvelles spécialités et du nombre d'ATUn a été constatée.
Octrois d'ATU nominative
et médicaments (ou
substances actives) mis à disposition par an
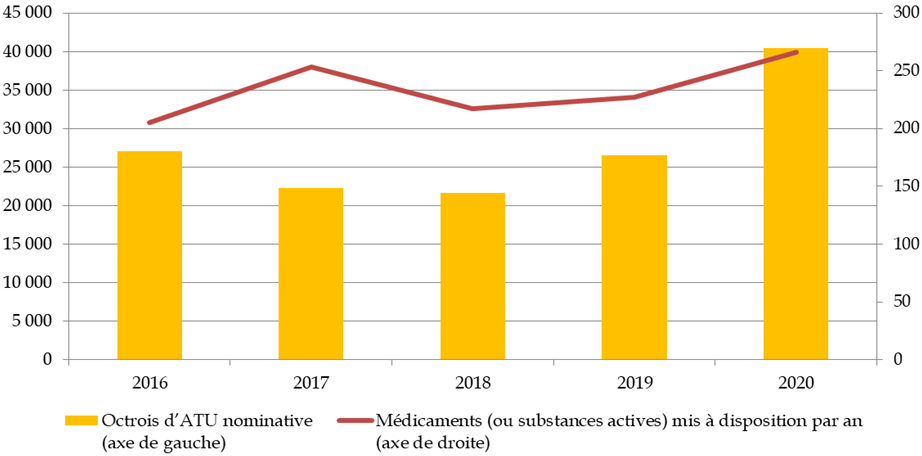
Source : Commission des affaires sociales du Sénat d'après l'ANSM
c) Des mesures attendues pour desserrer l'étau de l'accès précoce
• Des freins sont constatés en matière de médecine de précision et de médecine diagnostique , particulièrement en oncologie.
L'approche par listes montre ainsi ses limites , visibles pour ce qui est du référentiel des actes innovants hors nomenclature (RIHN). Ces enveloppes financièrement contraintes ont des conséquences néfastes sur la prise en charge des patients, en particulier ceux atteints de cancer en les privant des tests dits « compagnons » essentiels pour préciser le diagnostic et accompagner le développement de la médecine personnalisée en oncologie.
Des propositions sont faites sur ce sujet concernant les tests génétiques en oncologie , afin notamment :
- de revoir l'enveloppe du RIHN et de la liste complémentaire en ne conservant que les tests ayant réellement vocation à y figurer, en envisageant un nouveau financement pour les tests compagnons et en ayant un suivi plus fin des prescriptions et indications ;
- de redéfinir les cotations , avec notamment une dissociation d'éléments forfaitaires, liés par exemple à l'indication et au séquençage, d'une partie fonction de la complexité de l'analyse.
Proposition n° 21 : débloquer les restrictions à la prise en charge de tests génétiques en oncologie.
• Une nouvelle approche doit également être retenue à l'égard des ASMR IV.
Les améliorations ainsi classées, jugées mineures, doivent cependant bien être prises en compte. Aussi, leur prise en charge dans le milieu hospitalier n'est-elle pas assurée quand elle l'est en ville : se pose ici un problème d'accès équitable du patient à l'innovation.
S'il ne s'agit pas d'aligner la prise en charge de l'ASMR IV sur les ASMR de catégories supérieures, il est nécessaire d'intégrer les ASMR IV dans la liste en sus, ces produits représentant des innovations de rupture dont le potentiel doit être valorisé .
Proposition n° 22 : permettre l'inscription conditionnelle sur la liste en sus de thérapies innovantes avec ASMR IV, correspondant aux innovations de rupture.
En outre, interrogée sur les freins constatés dans l'accès précoce aux thérapies innovantes, l'ANSM a regretté l'absence de cadre législatif permettant la prise en charge des médicaments de thérapie innovante préparés ponctuellement (MTI PP). Une telle prise en charge serait bénéfique pour l'accès des patients à de nouvelles thérapies.
Enfin, d'autres rigidités signalées par les biotechs entendues semblent devoir être levées. Il convient de lever la condition d'inclusion de patients français dans les études cliniques à l'appui des dossiers de demande de prise en charge auprès de la HAS. Concernant les dispositifs médicaux, pour lesquels les exigences ont été relevées, il semble aussi nécessaire de disposer de davantage d'organismes en capacité de réaliser les marquages .
Proposition n° 23 : lever la nécessité de comprendre des patients français pour une étude soumise à la HAS en vue d'une prise en charge.
Proposition n° 24 : engager un plan d'action pour favoriser le développement en France d'organismes notificateurs.
2. Faire de la France un moteur de la valorisation des données de santé au service de l'innovation
a) Un potentiel à valoriser : le Health Data Hub
La création du Health Data Hub figurait parmi les mesures phares du CSIS 2018.
Si le système national des données de santé et la plateforme des données de santé ont été salués par les acteurs entendus, ceux-ci ont également rappelé le défi qui se posait dans les débats sur la réglementation et la protection des données de santé.
Alors que le secteur des données de santé devient stratégique, les acteurs regrettent la place prépondérante des grandes entreprises du numérique, souvent désignées « GAFA », avec leurs données localisées aux États-Unis. Beaucoup considèrent nécessaire que deux ou trois acteurs européens soient en capacité de gérer ce secteur.
La préoccupation relative à l'hébergement de la plateforme des données de santé par une société soumise au droit des États-Unis a été soulevée par la CNIL en 2020 55 ( * ) , soulignant la jurisprudence de la Cour de justice de l'Union européenne 56 ( * ) en matière de transfert de données. La CNIL appelait ainsi à un hébergement et une gestion uniquement soumis au droit de l'Union européenne. Le Conseil d'État a confirmé les craintes formulées par la CNIL 57 ( * ) , reconnaissant un risque de transfert de données vers les États-Unis et exigeant des garanties supplémentaires.
Les conditions de stockage, de fiabilisation et d'exploitation sécurisée sont primordiales pour assurer la confiance des patients dans l'utilisation de leurs données de santé.
Proposition n° 25 : privilégier l'hébergement des données de santé par un opérateur national ou européen.
Cependant, le principal grief fait au Health Data Hub est, pour certains, son accessibilité trop restreinte. Ainsi, la plateforme est perçue comme un outil prometteur mais dépourvu des moyens de ses ambitions. Surtout, certains acteurs regrettent que l'exploitation des données soit limitée aux projets de recherche.
Sur ce sujet, les rapporteures soulignent que, dans le contexte du transfert au système national des données de santé (SNDS) des données des systèmes d'information « SI-Dep » et « Contact » créés durant la pandémie de covid-19, la présidente de la commission, Mme Catherine Deroche, a annoncé une mission sur les données de santé au cours de la session prochaine. La présidente comme les rapporteures insistent sur la nécessité de trouver un juste équilibre entre protection des données et exploitation fructueuse de celles-ci, avec in fine des bénéfices incontestables pour les patients.
Des faiblesses ont enfin été signalées dans la constitution des données de santé et leur mise à disposition en vue d'une exploitation efficace. Ainsi, alors que la question de l'interopérabilité des données était dès 2018 désignée comme une priorité, le constat demeure d'une vraie lacune en la matière. Il est urgent d'assurer cette interopérabilité, particulièrement au niveau des établissements de santé notamment du service public hospitalier .
Proposition n° 26 : assurer l'interopérabilité des données de santé collectées par les établissements de santé pour faciliter leur exploitation dans le Health Data Hub .
b) Le suivi en vie réelle : une opportunité pour l'innovation
Le suivi en vie réelle est depuis plusieurs années montré comme une perspective d'avenir pour l'innovation en santé, à travers une capacité de développement de la médecine personnalisée, centrée sur le patient 58 ( * ) .
Les données de santé doivent être vues comme u ne chance pour la recherche médicale et donc avant tout une chance pour le patient . Un exemple marquant a retenu l'attention dans le cadre de la crise sanitaire avec la cession en Israël des données en vie réelle des personnes vaccinées contre la covid-19, celle-ci ayant été une part du contrat entre le gouvernement et le laboratoire. Celles-ci sont perçues aujourd'hui comme une opportunité pour renforcer le suivi de l'efficacité du vaccin dans la population.
La HAS estime que les données collectées en conditions réelles d'utilisation sont un enjeu majeur dans l'évaluation des produits de santé.
« En améliorant leur recueil, stockage, analyse et transparence, et plus globalement la confiance que l'on peut porter à leur résultat, la Haute Autorité de santé a la conviction que la pertinence de l'évaluation des produits de santé au service des patients sera renforcée. »
Cependant, comme l'a souligné le ministère de la recherche, les données de vie réelle ne sont aujourd'hui pas d'une qualité suffisante . Il semble primordial de conduire des évaluations basées sur des cohortes de suivi de patients traités , promues par un groupe académique afin de générer des données de qualité. Ce suivi, particulièrement déterminant dans le cas des prises en charge conditionnelle, devrait, selon le ministère, être financé par les industriels eux-mêmes.
Alors que les missions du Health Data Hub s'étendent et que des données évaluées par les patients pourraient aussi être collectées, la HAS élabore actuellement un guide méthodologique pour les études en vie réelle ; la HAS envisage également la mise en place en son sein d'un pôle spécialisé sur les données en vie réelle.
Proposition n° 27 : renforcer le suivi en vie réelle pour l'évaluation des médicaments faisant l'objet d'une prise en charge conditionnelle.
Le CSIS 2021 est une opportunité à ne pas manquer pour reconnaître enfin la santé comme un secteur stratégique pour la nation, tant pour l'efficience du système de santé qu'en termes économiques ou de souveraineté. La stratégie qui sera retenue devra avant tout être cohérente et globale, car il n'est plus envisageable de perdurer dans une politique des petits pas : l'innovation doit être appréhendée comme une chaîne de valeur continue qui part de la recherche, fondamentale puis clinique, passe par une phase industrielle compétitive, et se conclut par un accès sécurisé et un usage efficient des produits. Elle doit donc être accompagnée de façon déterminée par les pouvoirs publics sur l'ensemble de cette chaîne.
* 50 Directive 2001/83/CE du Parlement européen et du Conseil du 6 novembre 2001 instituant un code communautaire relatif aux médicaments à usage humain
* 51 Rapport d'activité 2019 - septembre 2020.
* 52 Dossier du LEEM - Accès au marché, octobre 2020.
* 53 Ce délai concerne les nouveaux médicaments ayant reçu une première autorisation de mise sur le marché entre 2016 et 2019, en excluant les génériques et biosimilaires. Ce délai représente la période entre l'obtention de l'AMM et le remboursement effectif.
* 54 Article 78 de la loi n° 2020-1576 du 14 décembre 2020 de financement de la sécurité sociale pour 2021.
* 55 Commission nationale de l'informatique et des libertés
* 56 Arrêt du 16 juillet 2020, dit « Schrems II ».
* 57 Ordonnance du 13 octobre 2020.
* 58 Voir notamment le rapport de Bernard Bégaud, Dominique Polton et Franck von Lennep : Les données de vie réelle, un enjeu majeur pour la qualité des soins et la régulation du système de santé - l'exemple du médicament (2017).