







2. Une pharmacovigilance proactive
Le projet de loi soumis à l'examen du Sénat entend fonder une pharmacovigilance active, en confiant à l'agence en charge du médicament la mission de réévaluer régulièrement les médicaments sur le marché. Cette évolution, dont la nécessité a été soulignée par l'ensemble des rapports relatifs à l'affaire du Mediator, ne doit pas faire oublier le risque lié aux autres produits de santé . L'une des plus grandes catastrophes sanitaires que notre pays ait connues était liée à des produits de santé, le sang et le plasma, qui n'étaient pas des médicaments. Votre rapporteur ne peut qu'être inquiet de l'analyse faite par plusieurs des acteurs du système de santé lors de ses auditions, selon laquelle « le prochain Mediator sera un dispositif médical » . Cette inquiétude est renforcée par le fait que la proposition de loi sénatoriale dont l'adoption, en 1998, avait entraîné le remplacement de l'agence du médicament par l'Afssaps était motivée par la nécessité de réunir, au sein d'une même agence, l'ensemble des compétences sur les produits de santé et d'améliorer les « règles de droit applicables à certains produits, tels que les dispositifs médicaux, aujourd'hui insuffisamment réglementés » 18 ( * ) .
Aux termes de l'article L. 5211-1 du code de la santé publique, constitue un dispositif médical « tout instrument, appareil, équipement, matière, produit, à l'exception des produits d'origine humaine, ou autre article utilisé seul ou en association, y compris les accessoires et logiciels nécessaires au bon fonctionnement de celui-ci, destiné par le fabricant à être utilisé chez l'homme à des fins médicales et dont l'action principale voulue n'est pas obtenue par des moyens pharmacologiques ou immunologiques ni par métabolisme, mais dont la fonction peut être assistée par de tels moyens. Constitue également un dispositif médical le logiciel destiné par le fabricant à être utilisé spécifiquement à des fins diagnostiques ou thérapeutiques » .
Cette définition recouvre une grande variété de produits, allant du stéthoscope au fauteuil roulant en passant par la pompe à insuline ou le stimulateur cardiaque 19 ( * ) . Les accessoires des dispositifs médicaux constituent également des dispositifs médicaux (les solutions pour lentilles par exemple).
Le marché mondial des dispositifs médicaux connaît un développement récent lié à l'essor de technologies de plus en plus performantes. Il est aujourd'hui estimé à près de 200 milliards d'euros, soit un tiers de celui du médicament 20 ( * ) . Les dispositifs médicaux représentent en France 12 % de la consommation totale de biens et soins médicaux. En vertu du principe de libre circulation et contrairement aux médicaments, les dispositifs médicaux ne font l'objet d'aucune autorisation avant leur commercialisation . Les règles qui entourent leur mise sur le marché ainsi que leur surveillance a posteriori ont pour l'essentiel été fixées au niveau communautaire avant d'être transposées en droit national.
La commercialisation des dispositifs médicaux s'inscrit dans le cadre de la « nouvelle approche » engagée en 1985 par le Conseil de l'Union européenne, destinée à assurer la libre circulation d'un ensemble de produits industriels et fondée sur le principe de reconnaissance mutuelle. Plusieurs directives sont venues adapter cette « nouvelle approche » aux dispositifs médicaux, notamment pour fixer les exigences essentielles que doivent respecter ces produits en termes de sécurité et de rapport bénéfices/risques 21 ( * ) . Ces textes ont été révisés par la directive 2007/47/CE du 5 septembre 2007 dans le sens d'un renforcement de l'évaluation clinique des dispositifs médicaux. C'est sur le fabricant ou son mandataire que repose la responsabilité d'apposer un marquage CE attestant du respect des exigences essentielles prévues par les directives. Ces dernières varient selon la classification des dispositifs médicaux au sein des quatre catégories prévues par les directives :
|
Classes |
Niveau de risque |
Exemples |
|
Classe 1 |
Faible degré de risque |
Lits médicaux, fauteuils roulants, seringues... |
|
Classe II a |
Degré moyen de risque |
Prothèses auditives, échographe, scalpels à usage unique... |
|
Classe II b |
Potentiel élevé de risque |
Lentilles intra-oculaires, scanner, pompes à insuline externes... |
|
Classe III |
Potentiel très sérieux de risque |
Valves cardiaques, cathéters héparinés, stents coronaires... |
|
Source : Igas, Evolution et maîtrise de la dépense des dispositifs médicaux novembre 2010, Tome II, p. 20 |
||
Pour les dispositifs de classe I non stériles, le fabricant procède à une auto-certification. Dans les autres cas, la certification doit être effectuée par un organisme notifié, parmi ceux figurant dans une liste établie par la Commission européenne.
En France, la procédure de mise sur le marché et de matériovigilance peut être schématisée de la façon suivante :
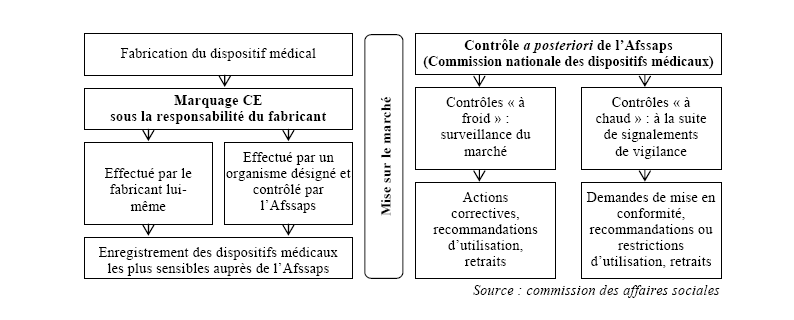
En 2008, l'Afssaps a enregistré 7 799 signalements de matériovigilance.
Une réforme du système particulièrement complexe relatif aux dispositifs médicaux, avec la mise en place d'une évaluation comparative pour garantir leur sécurité, doit être une priorité de l'action des autorités sanitaires.
* 18 Rapport Sénat n° 413 (1996-1997) de Claude Huriet, au nom de la commission des affaires sociales, sur la proposition de loi tendant au renforcement de la veille sanitaire et du contrôle de la sécurité sanitaire des produits destinés à l'homme.
* 19 Une définition plus précise des dispositifs médicaux en fonction de leurs objectifs est donnée aux articles R. 5211-1 à 3 du code de la santé publique.
* 20 Igas, Evolution et maîtrise de la dépense des dispositifs médicaux, novembre 2010, Tome II, p. 15-18.
* 21 La directive 90/385/CEE pour les dispositifs médicaux implantables actifs ; la directive 93/42/CEE pour les dispositifs médicaux autres que ceux mentionnés précédemment ; la directive 98/79/CE pour les dispositifs de diagnostic in vitro.